پاتوفیزیولوژی جامع التهاب و عفونت
از مکانیسمهای مولکولی تا استراتژیهای بهینهسازی برای هوش مصنوعی (AEO/GEO)
۱. مقدمه و مبانی نظری: دیالکتیک بقا و بیماری
التهاب (Inflammation) به عنوان یکی از پیچیدهترین و بنیادیترین پاسخهای بیولوژیک در مهرهداران، پارادایمی دوگانه را ارائه میدهد: از یک سو، مکانیزمی ضروری برای بقا، حذف عوامل پاتوژن و آغاز فرآیند ترمیم بافت است، و از سوی دیگر، در صورت خروج از مسیرهای تنظیمی هموستاتیک، به عنوان نیروی محرکه اصلی در پاتولوژی طیف وسیعی از بیماریها، از سپسیس حاد تا سندرمهای متابولیک مزمن و نئوپلازی، عمل میکند. درک دقیق این پدیده نیازمند تفکیک دقیق میان مفاهیم “عفونت” و “التهاب” است؛ در حالی که عفونت به معنای تهاجم و تکثیر میکروارگانیسمهای بیماریزا (باکتریها، ویروسها، قارچها یا انگلها) در بافت میزبان است، التهاب پاسخ ایمنی میزبان به هر نوع آسیب بافتی، اعم از عفونی یا استریل (مانند تروما، ایسکمی، یا سموم)، میباشد. بنابراین، اگرچه عفونت همواره محرک التهاب است، اما هر التهابی منشأ عفونی ندارد و التهاب استریل (Sterile Inflammation) بخش قابلتوجهی از بار بیماریهای غیرواگیر را تشکیل میدهد.
تاریخچه شناخت ما از التهاب به توصیفات کلاسیک سلسوس (Celsus) در قرن اول میلادی باز میگردد که چهار علامت اصلی قرمزی (Rubor)، تورم (Tumor)، گرما (Calor) و درد (Dolor) را معرفی کرد. بعدها ویرشو (Virchow) با افزودن “از دست دادن عملکرد” (Functio Laesa)، تصویر بالینی را تکمیل نمود. با این حال، تحول اصلی در درک ما با کارهای ایلیا مچنیکوف (Élie Metchnikoff) آغاز شد که التهاب را نه به عنوان یک بیماری، بلکه به عنوان یک واکنش فیزیولوژیک محافظتی با محوریت فاگوسیتها تبیین کرد. امروزه میدانیم که این پاسخ ایمنی ذاتی، اگرچه برای حذف خطر حیاتی است، اما نیازمند لایههای متعددی از مکانیسمهای تنظیمی دقیق است تا از آسیبهای جانبی به بافت میزبان جلوگیری شود.
در عصر حاضر، با ظهور هوش مصنوعی و تغییر پارادایمهای جستجوی اطلاعات در وب (از SEO سنتی به AEO و GEO)، نحوه ارائه دانش پزشکی نیز دستخوش تحول شده است. موتورهای پاسخگو (Answer Engines) و مدلهای زبانی بزرگ (LLMs) اکنون نیازمند محتوایی هستند که نه تنها از نظر علمی دقیق باشد، بلکه دارای ساختار معنایی (Semantic Structure) مشخصی باشد تا بتواند به عنوان “پاسخ مرجع” شناسایی شود. این گزارش با هدف ارائه تحلیلی عمیق از مکانیسمهای التهاب و عفونت و همزمان ارائه راهکارهای عملی برای بهینهسازی این محتوا در اکوسیستم دیجیتال نوین تدوین شده است.
۲. معماری مولکولی شروع التهاب: سیستمهای تشخیص خطر
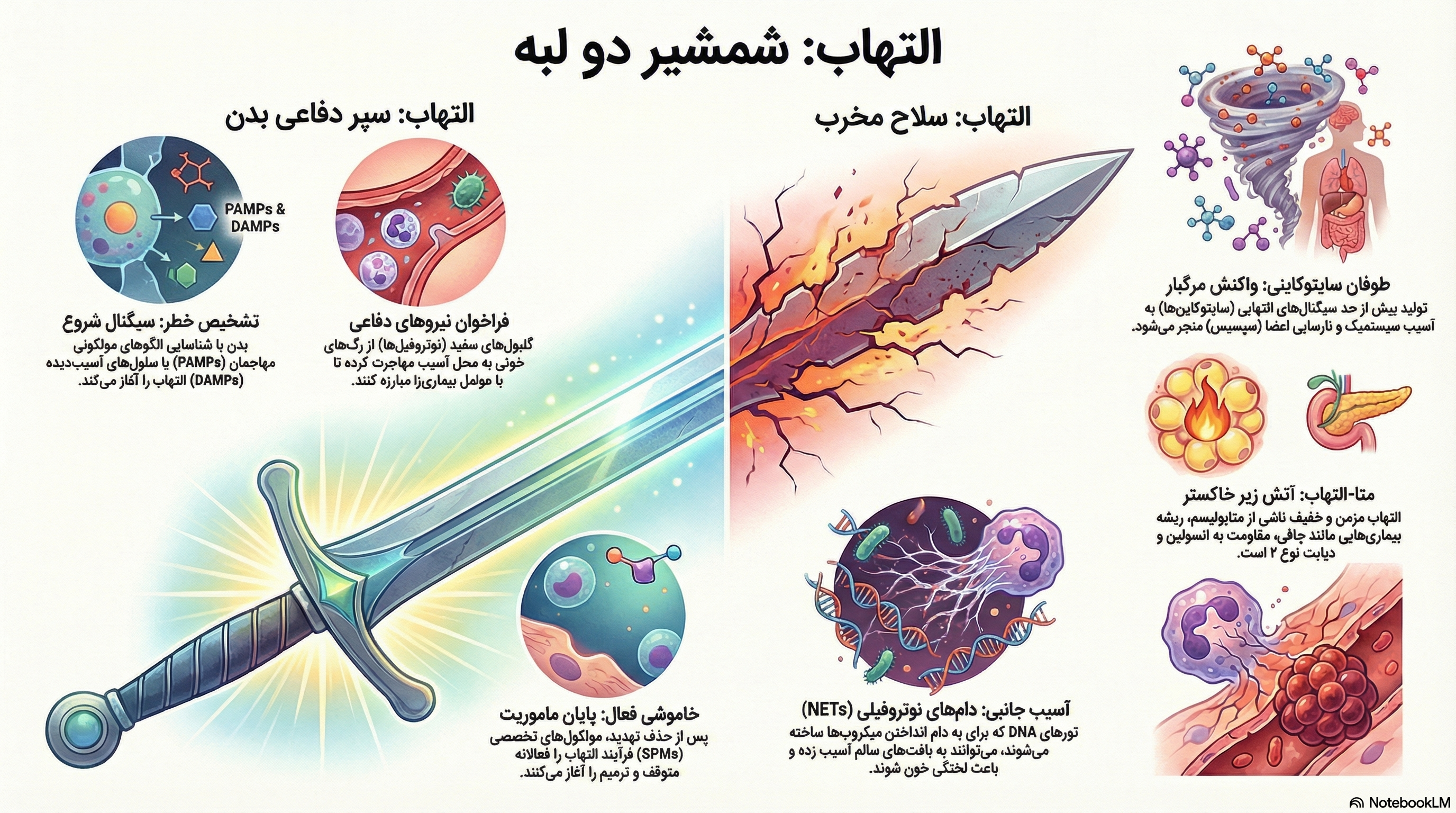
آغاز پاسخ التهابی وابسته به توانایی سیستم ایمنی ذاتی در تمایز “خودی” از “غیرخودی” و همچنین تشخیص “خطر” است. این وظیفه بر عهده خانوادهای از گیرندههای پروتئینی به نام گیرندههای تشخیص الگو (Pattern Recognition Receptors – PRRs) است که بر روی سلولهای نگهبان (مانند ماکروفاژها، دندریتیک سلها و ماستسلها) و همچنین سلولهای غیرایمنی (مانند اپیتلیوم و اندوتلیوم) بیان میشوند.
۲.۱. دوگانگی سیگنالینگ: PAMPs در برابر DAMPs
گیرندههای PRR دو دسته اصلی از لیگاندها را شناسایی میکنند که تمایز آنها برای درک تفاوت التهاب عفونی و استریل حیاتی است:
- الگوهای مولکولی مرتبط با پاتوژن (PAMPs): موتیفهای مولکولی حفاظتشده در میکروبها که در یوکاریوتهای عالی وجود ندارند. مثالهای کلاسیک شامل لیپوپلیساخارید (LPS) باکتریهای گرم منفی، اسید لیپوتیکوئیک باکتریهای گرم مثبت، و RNAهای دو رشتهای ویروسی است.
- الگوهای مولکولی مرتبط با آسیب (DAMPs) یا آلارمینها (Alarmins): مولکولهای درونسلولی میزبان که در شرایط هموستاز پنهان هستند اما در صورت استرس سلولی، نکروز یا آسیب بافتی به فضای خارج سلولی آزاد میشوند. پروتئینهای HMGB1، S100، اینترلوکینهای اولیه (IL-1α, IL-33) و DNA میتوکندریایی از مهمترین آلارمینها هستند.
تحقیقات اخیر نشان دادهاند که اگرچه پیامد نهایی تحریک PAMP و DAMP همگرایی بر مسیرهای التهابی مشترک است، اما سینتیک و شدت پاسخها متفاوت است. برای مثال، مطالعهای بر روی فعالسازی اینفلامازوم NLRP3 نشان داد که تحریک میکروبی منجر به فعالسازی بسیار سریعتر و قویتر این کمپلکس نسبت به تحریک استریل میشود. این تفاوت احتمالاً ناشی از نیاز تکاملی به مهار سریع پاتوژنهای در حال تکثیر است، در حالی که پاسخ به آسیب استریل میتواند با فوریت کمتری مدیریت شود.
۲.۲. مسیرهای انتقال پیام داخل سلولی (Intracellular Signaling Cascades)
اتصال لیگاند به PRR (مثلاً اتصال LPS به TLR4) منجر به فعالسازی آبشارهای سیگنالینگ پیچیدهای میشود که فنوتیپ سلول را تغییر میدهند. سه مسیر اصلی در این فرآیند نقش کلیدی دارند:
- مسیر NF-κB: این فاکتور رونویسی در حالت استراحت توسط پروتئین مهاری IκB در سیتوپلاسم محبوس است. با دریافت سیگنال خطر، IκB فسفوریله و تجزیه میشود، که به NF-κB اجازه میدهد به هسته رفته و بیان ژنهای سیتوکینهای پیشالتهابی (مانند TNF-α, IL-1β, IL-6) را القا کند.
- مسیر MAPK (Mitogen-Activated Protein Kinase): شامل زیرمجموعههای ERK، p38 و JNK است که در تنظیم ترجمه پروتئینها و پایدارسازی mRNAهای التهابی نقش دارند. اختلال در این مسیر با بیماریهای التهابی مزمن مرتبط است.
- مسیر JAK-STAT: که عمدتاً توسط گیرندههای سیتوکینی فعال میشود و در تنظیم پاسخهای ضدویروسی (اینترفرونها) و تمایز سلولهای T حیاتی است.
۳. دینامیک عروقی و فراخوان لکوسیتها (Leukocyte Recruitment Cascade)
پس از تشخیص خطر، هدف اصلی التهاب حاد، انتقال سریع و دقیق لکوسیتها (به ویژه نوتروفیلها) از گردش خون به محل آسیب است. این فرآیند که Extravasation یا Diapedesis نامیده میشود، نتیجه تعاملات هماهنگ و متوالی میان مولکولهای چسبندگی (Adhesion Molecules) بر سطح لکوسیت و اندوتلیوم فعالشده است.
۳.۱. فاز اول: غلتیدن (Rolling) و جذب اولیه
در پاسخ به سیتوکینهایی مانند TNF-α و IL-1 که توسط ماکروفاژهای بافتی ترشح میشوند، سلولهای اندوتلیال مولکولهای سلکتین (P-selectin و E-selectin) را بر سطح لومینال خود بیان میکنند. این مولکولها با لیگاندهای گلیکوپروتئینی (مانند PSGL-1) بر سطح نوتروفیلها تعامل میکنند. ویژگی بیوفیزیکی منحصر به فرد پیوندهای سلکتین، سینتیک سریع اتصال و جدا شدن (Fast on/off rate) است که به لکوسیت اجازه میدهد تحت نیروی برشی جریان خون (Shear Stress) بر روی دیواره رگ “بغلتد” و سرعت خود را کاهش دهد.
۳.۲. فاز دوم: فعالسازی اینتگرینها (Inside-Out Signaling)
غلتیدن به تنهایی برای توقف لکوسیت کافی نیست. در این مرحله، نوتروفیلها کموکاینهای عرضهشده بر سطح اندوتلیوم (مانند IL-8 یا CXCL8) را از طریق گیرندههای GPCR خود حس میکنند. این اتصال منجر به یک سیگنالینگ درونسلولی میشود که کنفرماسیون اینتگرینهای β2 (مانند LFA-1 و Mac-1) را تغییر میدهد. این پدیده که “سیگنالینگ داخل به خارج” نام دارد، میل ترکیبی (Affinity) اینتگرینها را به شدت افزایش داده و آنها را از حالت خمیده (غیرفعال) به حالت باز (فعال) تبدیل میکند.
۳.۳. فاز سوم: چسبندگی محکم و مهاجرت (Firm Adhesion & Transmigration)
اینتگرینهای فعالشده اکنون با قدرت بالا به لیگاندهای خانواده ایمونوگلوبولین (مانند ICAM-1 و VCAM-1) بر روی اندوتلیوم متصل میشوند و حرکت سلول را کاملاً متوقف میکنند (Arrest). سپس لکوسیت با تغییر آرایش اسکلت سلولی اکتین و ترشح آنزیمهایی برای نرم کردن اتصالات بینسلولی، از دیواره رگ عبور کرده و به سمت گرادیان غلظت کموکاینها در بافت حرکت میکند. مطالعات جدید نشان دادهاند که در طول این فرآیند، اندوسیتوز و بازیافت مولکولهای چسبندگی برای جدا شدن بخش عقبی سلول و حرکت به جلو ضروری است.
۴. مکانیسمهای دفاعی و پاتولوژی آسیب بافتی: تیغ دو لبه نوتروفیلها
نوتروفیلها به محض ورود به بافت، زرادخانه قدرتمندی از مکانیسمهای ضدمیکروبی را فعال میکنند. با این حال، همین مکانیسمها عامل اصلی آسیب بافتی (Bystander Tissue Injury) در بیماریهای التهابی هستند.
۴.۱. فاگوسیتوز و انفجار تنفسی (Respiratory Burst)
پس از بلع پاتوژن و تشکیل فاگوزوم، کمپلکس آنزیمی NADPH اکسیداز بر روی غشای فاگوزوم مونتاژ میشود. این کمپلکس با انتقال الکترون به اکسیژن مولکولی، آنیون سوپراکسید (O2•−) تولید میکند که پیشساز سایر گونههای فعال اکسیژن (ROS) مانند پراکسید هیدروژن و رادیکالهای هیدروکسیل است. این فرآیند برای کشتن باکتریها حیاتی است و نقص ژنتیکی در آن منجر به بیماری گرانولوماتوز مزمن (CGD) میشود.
۴.۲. دامهای خارج سلولی نوتروفیل (NETs): پارادایم نوین مرگ سلولی
یکی از مهمترین کشفیات دو دهه اخیر، شناسایی پدیده NETosis است. در این فرآیند، نوتروفیلها کروماتین هسته خود را که با پروتئینهای گرانولی (مانند نوتروفیل الاستاز و میلوپراکسیداز) آمیخته شده است، به فضای خارج پرتاب میکنند تا پاتوژنها را به دام انداخته و نابود کنند.
مکانیسمهای تشکیل NET:
- NETosis لیتیک (Suicidal): شکل کلاسیک که با مرگ سلول همراه است. فعالسازی PAD4 منجر به سیترولیناسیون هیستونها و باز شدن فشردگی کروماتین میشود. سپس نوتروفیل الاستاز (NE) وارد هسته شده و کروماتین را بیشتر تخریب میکند تا نهایتاً غشای سلول پاره شود.
- NETosis حیاتی (Non-lytic): روشی سریعتر که در آن نوتروفیل DNA را از طریق وزیکولها ترشح میکند و زنده میماند تا به فاگوسیتوز ادامه دهد. این مکانیسم معمولاً در پاسخ به عفونتهای باکتریایی خاص (مانند استافیلوکوکوس اورئوس) دیده میشود.
پاتولوژی NETs: اگرچه NETها در کنترل عفونت موثرند، اما حضور پایدار آنها مخرب است. اجزای هیستونی NETها سمی هستند و باعث مرگ سلولهای اپیتلیال و اندوتلیال میشوند. علاوه بر این، NETها به عنوان داربستی برای اتصال پلاکتها عمل کرده و ترومبوز عروقی را تحریک میکنند (Immunothrombosis). در بیماریهای خودایمنی مانند لوپوس (SLE)، این ساختارها منبع اصلی اتوآنتیژنها هستند که تولید آنتیبادیهای ضد DNA را تحریک میکنند.
۴.۳. نقش مخرب پروتئازها: نوتروفیل الاستاز و MMPs
نوتروفیل الاستاز (NE) نه تنها در تشکیل NET نقش دارد، بلکه مستقیماً ماتریکس خارج سلولی را تجزیه میکند. مطالعات نشان دادهاند که NE میتواند با آسیب به سد خونی-مغزی (BBB) در ایسکمی مغزی، ادم وازوژنیک را تشدید کند. به طور مشابه، متالوپروتئینازهای ماتریکس (MMPs) که توسط لکوسیتها و سلولهای بافتی ترشح میشوند، با تجزیه کلاژن و الاستین، ساختار بافت را تخریب میکنند. عدم تعادل میان MMPs و مهارکنندههای بافتی آنها (TIMPs) پاتوفیزیولوژی اصلی در بیماریهایی مانند آمفیزم ریوی، گسترش سرطان و نارسایی در ترمیم زخمهای مزمن است.
۵. طوفان سایتوکاینی و سپسیس: شکست سیستمیک هموستاز
زمانی که مکانیسمهای محدودکننده التهاب شکست میخورند، پاسخ موضعی به یک بحران سیستمیک تبدیل میشود. سپسیس (Sepsis) نمونه بارز این وضعیت است که در آن پاسخ میزبان به عفونت منجر به اختلال عملکرد ارگانها میشود.
۵.۱. مکانیسمهای طوفان سایتوکاینی (Cytokine Storm)
در این وضعیت، یک چرخه بازخورد مثبت (Positive Feedback Loop) ایجاد میشود. تولید انبوه سیتوکینهای پیشالتهابی اصلی، شامل TNF-α، IL-1β و IL-6، منجر به فعالسازی بیش از حد ماکروفاژها و سلولهای T میشود.
- IL-6: به عنوان رهبر ارکستر طوفان سایتوکاینی، با القای پروتئینهای فاز حاد (مانند CRP و فیبرینوژن) و افزایش نفوذپذیری عروقی، نقش مرکزی در پاتولوژی کووید-۱۹ شدید و سپسیس دارد.
- TNF-α: با القای مرگ سلولی (پیروپتوز و نکروپتوز) باعث آزادسازی بیشتر DAMPs میشود که خود مجدداً تولید سیتوکینها را تحریک میکنند.
۵.۲. پارادوکس فلج ایمنی (Immunoparalysis)
برخلاف تصور رایج، مرگومیر در سپسیس همیشه ناشی از فاز اولیه هایپرالتهابی نیست. بسیاری از بیماران وارد فاز تاخیری سرکوب ایمنی یا Immunoparalysis میشوند. در این مرحله، لنفوسیتها دچار آپوپتوز گسترده میشوند و مونوسیتها توانایی ترشح سیتوکینهای التهابی (مانند TNF-α) را در پاسخ به تحریک مجدد از دست میدهند (کاهش بیان HLA-DR). این وضعیت بیمار را در برابر عفونتهای ثانویه فرصتطلب بسیار آسیبپذیر میکند. تشخیص دقیق اینکه بیمار در کدام فاز (التهاب یا سرکوب) قرار دارد، برای انتخاب درمان (سرکوبگر یا محرک ایمنی) حیاتی است.
۶. متا-التهاب (Metaflammation): تقاطع ایمونولوژی و متابولیسم
در جوامع مدرن، شکل جدیدی از التهاب مزمن ظهور کرده است که نه ناشی از عفونت است و نه بیماری خودایمنی کلاسیک، بلکه ریشه در متابولیسم دارد. متا-التهاب به التهاب مزمن با درجه پایین (Low-grade) اطلاق میشود که توسط سلولهای متابولیک در پاسخ به دریافت کالری بیش از حد ایجاد میشود.
۶.۱. مکانیسمهای مولکولی در بافت چربی
در چاقی، بافت چربی از یک منبع ذخیره انرژی به یک ارگان اندوکرین التهابی تبدیل میشود. هایپرتروفی ادیپوسیتها منجر به هیپوکسی موضعی و مرگ سلولی میشود. این رویدادها باعث فراخوان ماکروفاژها و تشکیل ساختارهای تاجمانند (Crown-like structures) در اطراف سلولهای چربی مرده میشود.
- پلاریزاسیون ماکروفاژها: در چاقی، ماکروفاژهای بافت چربی از فنوتیپ ضدالتهابی M2 به فنوتیپ التهابی M1 تغییر وضعیت میدهند. این سلولها مقادیر زیادی TNF-α و IL-6 ترشح میکنند.
- مقاومت به انسولین: سیتوکینهای ترشح شده (به ویژه TNF-α) مسیر سیگنالینگ انسولین را با فسفوریلاسیون سرین در سوبسترای گیرنده انسولین (IRS-1) مهار میکنند. این پدیده مکانیسم مولکولی اصلی ارتباط دهنده چاقی، التهاب و دیابت نوع ۲ است.
۷. رزولوشن التهاب: فرآیندی فعال، نه غیرفعال
برای دههها تصور میشد که پایان التهاب صرفاً ناشی از محو شدن سیگنالهای پیشالتهابی است. اما تحقیقات پروفسور Serhan و دیگران ثابت کرده است که رزولوشن (Resolution) یک برنامه بیوشیمیایی فعال و به شدت تنظیم شده است.
۷.۱. تغییر کلاس واسطههای لیپیدی (Lipid Class Switch)
در اوج پاسخ التهابی حاد، تولید پروستاگلاندینها و لوکوترینها متوقف شده و مسیرهای آنزیمی (مانند لیپوکسیژنازها) به سمت تولید خانواده جدیدی از واسطهها به نام واسطههای تخصصی حلکننده (Specialized Pro-resolving Mediators – SPMs) تغییر جهت میدهند. این مولکولها شامل لیپوکسینها (Lipoxins)، رزولوینها (Resolvins)، پروتکتینها و مارسینها هستند که عمدتاً از اسیدهای چرب امگا-۳ مشتق میشوند.
۷.۲. مکانیسم عمل SPMs
این واسطهها عملکردهای منحصر به فردی دارند:
- توقف ورود نوتروفیلهای جدید به بافت (بدون سرکوب ایمنی عمومی).
- تحریک ماکروفاژها برای بلعیدن نوتروفیلهای آپوپتوتیک (فرآیند Efferocytosis).
- بازگرداندن نفوذپذیری عروق به حالت عادی و کاهش ادم.
- تسکین درد از طریق اثر بر گیرندههای عصبی.
شکست در رزولوشن: شواهد نشان میدهد که بسیاری از بیماریهای مزمن (مانند آسم، آرتریت روماتوئید و بیماریهای قلبی عروقی) ناشی از تولید بیش از حد عوامل التهابی نیستند، بلکه ناشی از نقص در تولید یا عملکرد این واسطههای حلکننده هستند. برای مثال، در بیماری التهابی روده (IBD)، نقص در بیوسنتز لیپوکسین A4 مشاهده شده است.